

Drug Hub: Sophisticated Machine Learning Algorithms to Address Pharmaceutical Needs
The process of discovering and designing new drugs is complex, expensive, and time-consuming. Developing a new drug is a multistage process that takes fifteen years on average. Different drugs may have their own unique requirements, but the overarching process consists of: discovery, preclinical testing, clinical trials, review, and approval.
Today, pharmaceutical companies must invest a major portion of their time and resources into the preclinical, R&D, stage. As a result, pharmaceutical companies are constantly looking for opportunities to reduce the costs, time, and risks of this preclinical phase. In the discovery phase, which consumes about $600 MM and takes about seven years per drug, researchers identify drug candidates by searching for compounds that can fit into a specific cavity, or bind with, a protein target.
Traditional High Throughput Screening
High Throughput Screening (HTS) is the classic experimental approach for drug development that is physically carried out in labs. Researchers start with a specific target protein and thousands (or even millions) of compounds or molecules of interest. A robot then automatically prepares test tubes with the target protein and one possible compound. Then, researchers must wait several days for a reaction to occur and manually check to see if the compound bonded to the protein. As you can imagine, this is very time consuming and expensive. A virtual version of HTS cannot replace the physical test entirely, but it can reduce the amount of physical testing required by predicting the most viable candidate substances for subsequent screening.
The Big Data Solution: Virtual HTS
The virtual screening process, also known as in silico screening, uses predictive models to narrow down the number of candidate molecules. Fundamentally, virtual screening predicts the likelihood that an arbitrary molecule will bind with the target protein. After the virtual screening has identified a small number of compounds as drug candidates, traditional HTS can be done to perform final preclinical testing with the reduced number of candidate molecules.
By combining a scalable data platform with leading machine learning algorithms, Virtual HTS can help evolve traditional HTS in exciting new ways. Virtual HTS reduces costs for the pharmaceutical industry by shortening drug design research cycles, calculating results more quickly, and making results more accurate, meaning we can find the best-fitting compounds more precisely than in the classic approaches.
Virtual HTS in Detail
The Data
The Virtual HTS process begins with a database of molecules—potentially every molecule you can imagine. For example, the open database PubChem contains more than sixty million chemicals. To solve any prediction problem like this one, we must calculate variables. In the case of Virtual HTS, these variables describe the molecule’s physical and chemical properties, such as molecular weight, number of atoms, number of carbon atoms, total charge, and number of rings. The Dragon 7 package, the standard package for molecular descriptors, contains these and 5,265 other molecular descriptors.
The screening data, which contains billions of results from thousands of historic screening runs generated in various labs over the years, is an even larger dataset. Every data point is valuable not only because of the information it can provide but also because it was generated through expensive physical HTS runs. A scalable open data platform like Opera’s is invaluable because it allows the user to integrate all of these molecular descriptors and historic screening data in one place. This results in a massive dataset, which is perfect for solving a supervised learning task like drug discovery. We can use models, such as deep neural networks or boosted decision trees, to predict the target bindings and rank molecules according to their likelihood of success. Of course, the results from the executed HTS runs are ultimately fed back into the screening database to help improve subsequent predictions.
Using the Data
Supervised learning is a machine learning methodology that requires the model to learn from labeled training data, or data for which the dependent variable outcome has been marked. This is the approach we can use to solve the Virtual HTS problem because we do have a good amount of labeled data. What exactly does this mean in practice? Let’s take a look at the matrix below to help us visualize the data better:
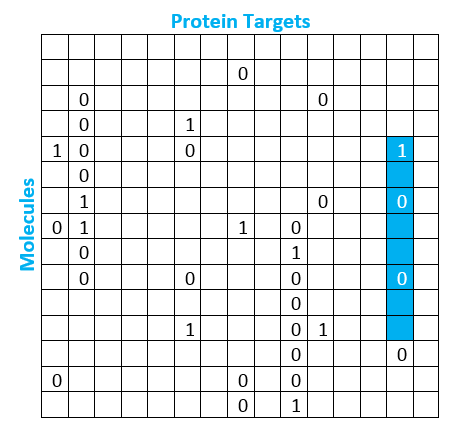
This matrix helps us visualize the available data around the relationship between molecules and protein targets from past screening runs. In reality, this matrix is massive, containing millions of molecule rows and tens of thousands of protein target columns. A box containing a 1 indicates positive results in the screening data, meaning the molecule bonded to the protein target. A box containing a 0 indicates negative results in the screening data, meaning the molecule did not bind to the protein target. As we would expect, the actual matrix contains far fewer ones than zeroes. Less than 1% of the observations are positive, creating an imbalanced dataset. However, most of the boxes are actually empty, which means we have no historic observation of whether or not that molecule binds to that protein. We have highlighted in blue the column that represents the outcome for several molecules for our protein target of interest.
Solving the Problem
Virtual HTS is a classification problem, which means we are trying to predict a label rather than trying to predict a specific quantity. In our case, we have established that we are trying to predict the label 0 or 1 to indicate whether or not a molecule will bind to our protein target. Let’s take a look at the matrix below to understand what this looks like as a binary classification problem:
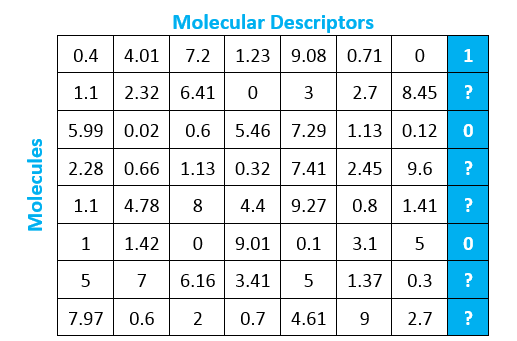
Each row represents a molecule, and each column represents a variable representation of a molecular descriptor. The blue column is the same as the blue column from the earlier matrix. This variable - whether or not the molecule will bind with the protein of interest - is what we are trying to predict using the other molecular descriptors.
The rows for which the blue column is populated can be used as training data, while the rows for which the blue column remains unpopulated can be used for testing. In other words, we let the model learn from the rows that tell us whether or not the molecule bonded with the protein so that the model can predict the likelihood that the other molecules (for which we do not have empirical evidence) will bind to the protein. The predicted outcome for these unknown molecules helps us narrow down the molecules for which we must perform physical experiments using traditional HTS.
Benefits for the Pharmaceutical Industry
For pharmaceutical companies, there are many benefits from solutions that combine Big Data capabilities and sophisticated deep learning algorithms. As Big Data capabilities continue to improve, they will enable Virtual HTS, thereby providing significant performance boosts over classic drug discovery approaches in terms of costs and time.
At Opera Solutions, we take our AI solutions to the next level, exploring novel applications of our technology across diverse fields. Interested in learning more? Click here to learn more about our approach to practical AI or drop us a note!